Abstract
Transthyretin amyloid cardiomyopathy (ATTR-CM) is an increasingly recognized cause of heart failure in older adults. Advances in molecular pharmacotherapy have transformed the therapeutic landscape, shifting focus from symptomatic management to disease-modifying interventions.
Introduction
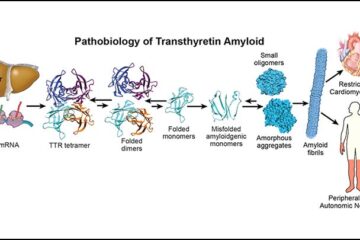
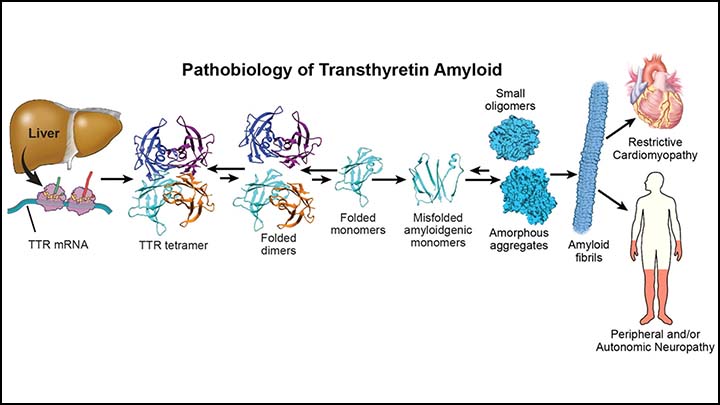
ATTR-CM results from the deposition of misfolded transthyretin protein in myocardial tissue, leading to progressive restrictive cardiomyopathy. Historically underdiagnosed, improved awareness and imaging have contributed to earlier identification.
Molecular Pathophysiology
Disease progression involves:
- Transthyretin tetramer destabilization
- Protein misfolding and aggregation
- Amyloid fibril deposition in cardiac tissue
Targeting these molecular steps has become central to modern treatment strategies.
Emerging Pharmacological Targets
Recent advances focus on:
- Stabilization of transthyretin tetramers
- Reduction of transthyretin production
- Promotion of amyloid clearance
These approaches represent a paradigm shift from supportive care to targeted therapy.
Clinical Implications
Molecular therapies have demonstrated improvements in functional capacity, quality of life, and disease progression markers. Early diagnosis is now critical to maximizing therapeutic benefit.
Conclusion
The evolution of molecular-targeted therapies marks a significant milestone in the management of transthyretin amyloid cardiomyopathy. Ongoing research continues to refine treatment strategies and long-term outcomes.